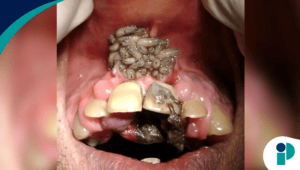
La fibrodysplasia ossificans progressiva (FOP), también conocida como «enfermedad del hombre de piedra» o «enfermedad de Münchmeyer«, es una condición congénita extremadamente rara que provoca la formación de un segundo esqueleto en el cuerpo humano, dejando a los pacientes inmovilizados. Esta patología, que afecta a aproximadamente 1 de cada 1 millón de personas, no discrimina por sexo, raza o etnicidad, y sus implicaciones médicas y sociales son profundas.
La genética lo implica todo en la fibrodysplasia ossificans progressiva
La FOP es causada por una mutación en el gen ACVR1, que juega un papel crucial en la formación del esqueleto durante el desarrollo embrionario y en la reparación ósea a lo largo de la vida. Esta mutación provoca una hiperactividad del gen, lo que desencadena la formación descontrolada de hueso en tejidos donde no debería existir, como músculos, ligamentos y tendones.
En la mayoría de los casos, esta mutación ocurre de manera aleatoria, sin antecedentes familiares de la enfermedad. Sin embargo, en raras ocasiones, puede heredarse de un progenitor afectado, siendo suficiente una sola copia del gen mutado para que se desarrolle la condición.
Una mutación del gen que coordina la formación adecuada del esqueleto
El gen ACVR1, también conocido como receptor activin tipo 1, es clave en la regulación del desarrollo óseo y el mantenimiento del esqueleto. Este gen codifica una proteína que forma parte de la familia de receptores de las proteínas morfogenéticas óseas (BMP, por sus siglas en inglés), responsables de procesos celulares como la formación del hueso y la reparación de tejidos tras una lesión. Durante el desarrollo embrionario, el ACVR1 coordina la formación adecuada del esqueleto, mientras que en la vida adulta, regula la regeneración ósea de manera controlada. Sin embargo, una mutación en este gen puede hiperactivar su función, causando la formación anómala de hueso en tejidos blandos, como ocurre en la fibrodysplasia ossificans progressiva (FOP). Esta actividad descontrolada interfiere con la señalización normal, desencadenando una osificación ectópica progresiva y debilitante.
¿Cuáles son las manifestaciones clínicas de la enfermedad del hombre de piedra?
Desde el nacimiento, uno de los signos más característicos de la FOP es la presencia de dedos gordos de los pies cortos y volteados hacia adentro. Alrededor del 50% de los pacientes también presenta deformaciones en los pulgares. Sin embargo, los síntomas más debilitantes suelen aparecer durante la infancia, cuando comienzan los episodios de osificación progresiva.
Las áreas más comúnmente afectadas al inicio incluyen el cuello, la espalda, el pecho y las extremidades. Durante los llamados «brotes» o flare-ups, los pacientes experimentan inflamación, dolor y rigidez, lo que acelera el proceso de osificación. Estos brotes pueden desencadenarse espontáneamente o debido a traumatismos, como lesiones, cirugías o infecciones virales. Además, las complicaciones incluyen dificultades para hablar o comer si la mandíbula se ve afectada, pérdida de audición y deformidades espinales.
Con el tiempo, el crecimiento de hueso extra en tejidos blandos genera un segundo esqueleto que limita severamente la movilidad. Para los 30 años, la mayoría de los pacientes pierden por completo la capacidad de moverse. La expectativa de vida promedio es de 56 años, y la causa más común de muerte es la insuficiencia cardiorrespiratoria debido a la imposibilidad de respirar correctamente.
Avances en el díficil tratamiento de la fibrodysplasia ossificans progressiva
Aunque no existe una cura para la FOP, los esfuerzos se centran en el manejo de los síntomas y la prevención de brotes. Los analgésicos y los medicamentos antiinflamatorios, como los corticosteroides, son útiles para aliviar el dolor y la inflamación. También se recomienda que los pacientes eviten lesiones e infecciones para minimizar los riesgos de osificación. La cirugía, aunque podría parecer una solución para eliminar el hueso adicional, está contraindicada, ya que puede agravar la formación ósea.
Un avance significativo ocurrió en 2023, cuando la Administración de Alimentos y Medicamentos de Estados Unidos (FDA) aprobó el uso de un medicamento oral llamado palovaroteno (Sohonos). Este fármaco, indicado para niñas mayores de 8 años y niños mayores de 10, mostró resultados prometedores en ensayos clínicos. En un estudio con 107 pacientes, aquellos tratados con palovaroteno experimentaron una reducción del 54% en la formación excesiva de hueso en comparación con los grupos de control. Este hito marca un paso adelante en la mejora de la calidad de vida de los pacientes, aunque el tratamiento no detiene por completo la progresión de la enfermedad.
¿Qué trae el futuro para los pacientes con Fibrodisplasia osificante progresiva?
Los estudios actuales se centran en comprender mejor los mecanismos moleculares de la FOP y desarrollar terapias genéticas que puedan silenciar o corregir la mutación en el gen ACVR1. Investigaciones en modelos animales han demostrado que la manipulación de vías celulares relacionadas con la mutación podría ser efectiva. Además, tecnologías como la edición genética CRISPR ofrecen esperanzas a largo plazo para modificar directamente el ADN defectuoso.
Otra área de interés es la identificación de biomarcadores que permitan predecir brotes y evaluar la eficacia de nuevos tratamientos. Los investigadores también están explorando terapias combinadas que utilicen medicamentos existentes junto con nuevas moléculas en desarrollo.
Investigación científica ofrece esperanza a una condición rara y grave en los humanos
La fibrodysplasia ossificans progressiva representa un desafío médico y científico único debido a su rareza y gravedad. Aunque los avances recientes en tratamientos, como el palovaroteno, son prometedores, aún queda mucho por descubrir para mejorar las perspectivas de los pacientes. La investigación continua y la colaboración internacional serán esenciales para encontrar una cura y, mientras tanto, brindar un apoyo integral a quienes enfrentan esta devastadora enfermedad